In June 2022, the Food and Drug Administration (FDA) published a Guidance document1 to serve manufacturers of radiological devices as guidance when filling in their premarket submissions for such devices if they include quantitative imaging functions.
We have summarized this guidance to give manufacturers and radiologists a clear overview of the Guidance’s content and the impact is may have on the industry.
FDA guidance for Technical Performance Assessment of Quantitative Imaging in Radiological Device Premarket Submissions
Quantitative imaging values, as output from medical devices that include quantitative imaging functions, are usually subject to both systematic error and random variation. Thus, a quantitative imaging value can, and usually does, differ from the true value of the measurand. Errors may come from the acquisition of the medical images, patient characteristics, and the image processing algorithm. An understanding of the sources of error, especially those with the largest impact on the measurand and the quantitative imaging values produced by the quantitative imaging function, is important for characterizing the performance of the quantitative imaging function/device.
Therefore, the premarket submission should provide performance specification, supporting performance data to demonstrate that the quantitative imaging values meet the performance specifications, and information for the end user to understand and interpret the values provided by the device.
As is the case with every FDA Guidance document, the guidance content provided by the FDA for this matter is not legally binding. However, the document contains recommendations on what the FDA considers best practice, and therefore on what is expected to be addressed in a pre-market submission.
For that reason, this FDA Guidance for Technical Performance Assessment of Quantitative Imaging in Radiological Device Premarket Submissions1 provides very useful information for manufacturers of radiological devices that include quantitative imaging functions independently of the devices intended uses, the level of automation, the complexity of their algorithms or their imaging modality. Particularly, the Guidance addresses recommendations related to the description, user-information and performance assessment that are normally included in the pre-market submissions.
Scope
The guidance, as per FDA’s indication, can be applied to a large number of premarket submission types, including premarket notification (510(k)) submissions, De Novo requests and premarket approval (PMA) applications.
Of course this does not replace other Guidance documents on the content of a premarket submission for software, as described for example in other FDA references2,3 and FDA’s device specific guidances or special controls.
It is important to note that the clinical evaluation of software as a medical device is explicitly not included in the scope of this Guidance as this specific type of software has its own guidance document4.
Potential sources of measurement error
Quantitative imaging extracts additional information from medical images in the form of numerical values. These values are usually subject to both systematic error, and random variation. Since these sources of error have an impact on the reported outputs, they may influence clinical decision-making.
The Guidance1 provides a clear description of what sources of systematic error and random variation may affect quantitative imaging values, or medical imaging data. It includes a list of potential sources, divided in:
- Patient characteristics (physiology, movement of tissue, differences in physiological state etc.)
- Image acquisition (patient positioning, imaging hardware and protocol, artifacts, etc.)
- Image processing (algorithm specifics, seed point selection, curve fitting etc.)
Although systematic errors and random variation cannot be prevented due to the nature of the subjects (living patients) and the imaging acquisition and processing actions, quantitative imaging values are of greatest worth when the performance of the imaging function is well characterized. This means that users have sufficient information to understand and interpret the values being reported.
What to include in a Premarket Submission of a radiological device that include a quntitative imaging function
There are different sets of documentation that accompany medical devices. The premarket submission documentation contains numerous documents for reviewing purposes, from which only a limited subset is intended to be information provided to the End User.
According to the FDA guidance1, if you manufacture a radiological device that includes a quantitative imaging function, you should include the following information in your premarket submission:
- Function description: “a technical description of the quantitative imaging function(s) included in the device”. This description should also contain details referring to the implementation of the algorithm’s software according to previous FDA’s recommendations3.
- Technical Performance Assessment: quantitative performance specifications and data to corroborate the device’s claims and uncertainty regarding the imaging function described.
- Labeling (User Instructions): information to ensure that the user can obtain, understand and interpret the output of the imaging function appropriately. The labeling included must satisfy all FDA’s labeling requirements that might be applicable.
This End User information, also known as Labeling or User Instructions, is part of the premarket submission, but warrant’s its own section, since it is this labeling documentation that provides the user information on how to understand and interpret the values.
Function Description
This information is directed at the reviewer of the submission. Based on the recent FDA recommendations1, when describing the quantitative imaging function of a radiological device manufacturers should include the description of the quantitative imaging function itself, including the following details:
- The measurand
- The software platform, name, version or characteristics
- The algorithm used, its inputs and outputs
- The level of automation of the imaging function, whether it is manual, semi-automatic or automatic
- The input data used, details on its restrictions, limitations and target populations
- The processes to ensure image acceptance, which means detailing how does the manufacturer ensure that input data is acceptable for its processing by the algorithm, and if that is done automatically or not
- The information and instructions given to the end user
- The level of user interaction that the device needs for intended use
Technical Performance Assessment
The premarket submission should include performance specifications for the quantitative imaging functions, preferably with objective reference values, that will depend on characteristics such as the intended use, the complexity of the measurement algorithm, and the availability of reference values. Moreover, these specifications are susceptible to change throughout the operation range of the imaging function. Supporting performance data is needed to prove that the device meets the predefined performance specifications.
Recommended steps1 to follow when filing the technical performance assessment of the device’s quantitative imaging function:
- Describe the imaging function, its relationship to the measurand and the conditions of use.
- Set the reference standard and the applicable performance metrics for the device (generally including metrics such as uncertainty, sensitivity, specificity, precision or bias).
- Specify the performance of the imaging function under the conditions set in the description of the device.
- Define the experimental unit, the statistical estimates of performance (including the limits of agreement for example), and the acceptance criteria or goals based on its intended use and limitations.
- State the elements of the statistical design (including the needed data and the analytical plan).
- Collect the necessary data, run the statistical analysis and compare its results to the acceptance criteria previously defined.
The manufacturer’s claims about the device’s imaging function performance should be based on studies that have pre-defined acceptance criteria. If there are claims about the imaging function performing better than manual methods, these claims should be supported by studies that compare automated outputs to outputs derived manually from a group of experts1.
Labeling (User instructions)
The manufacturer should include labeling in their pre-market submission. The information included aims to ensure the appropriate use of the imaging function and the device and as such, should include1 a description of the measurand and the algorithm inputs; details on performance specifications and uncertainty, if uncertainty cannot be measured for the imaging function the manufacturer has to provide potential sources of output variability; instructions for image acceptance processes the user needs to perform; a description of the training and the qualifications needed for the user to perform the intended use of the device; and, if the imaging function compares the outputs to a reference database, the manufacturer should provide information on the configuration of said database.
Are you interested in AI radiology software? Check out our 101 guide to its FDA regulatory process.
What to include in manual, semi-automated and fully automated quantitative imaging functions premarket submissions
Manual quantitative imaging functions
For radiological devices in which physicians make quantitative measurements using a fully manual function, the function itself should be completely transparent in order to enable users to estimate whether the device is useful for their clinical needs. It should also be clear if the function has had any type of validation.
The FDA guidance recommends1 the premarket submission for radiological devices with manual quantitative imaging functions to include:
- Function description: it should describe functionality of the quantitative imaging function in depth and clearly specify if and what algorithms are implemented in the function.
- Technical Performance Assessment: it should include details about the software verification activities that ensure its correct implementation. Although performance claims might not be included for this type of function, if added, they should be supported.
- Labeling (user interaction): it should include a clear description of the function’s functionality, any limitations on input images and any performance specifications or its absence. For the latter case, sources of output variability should also be included.
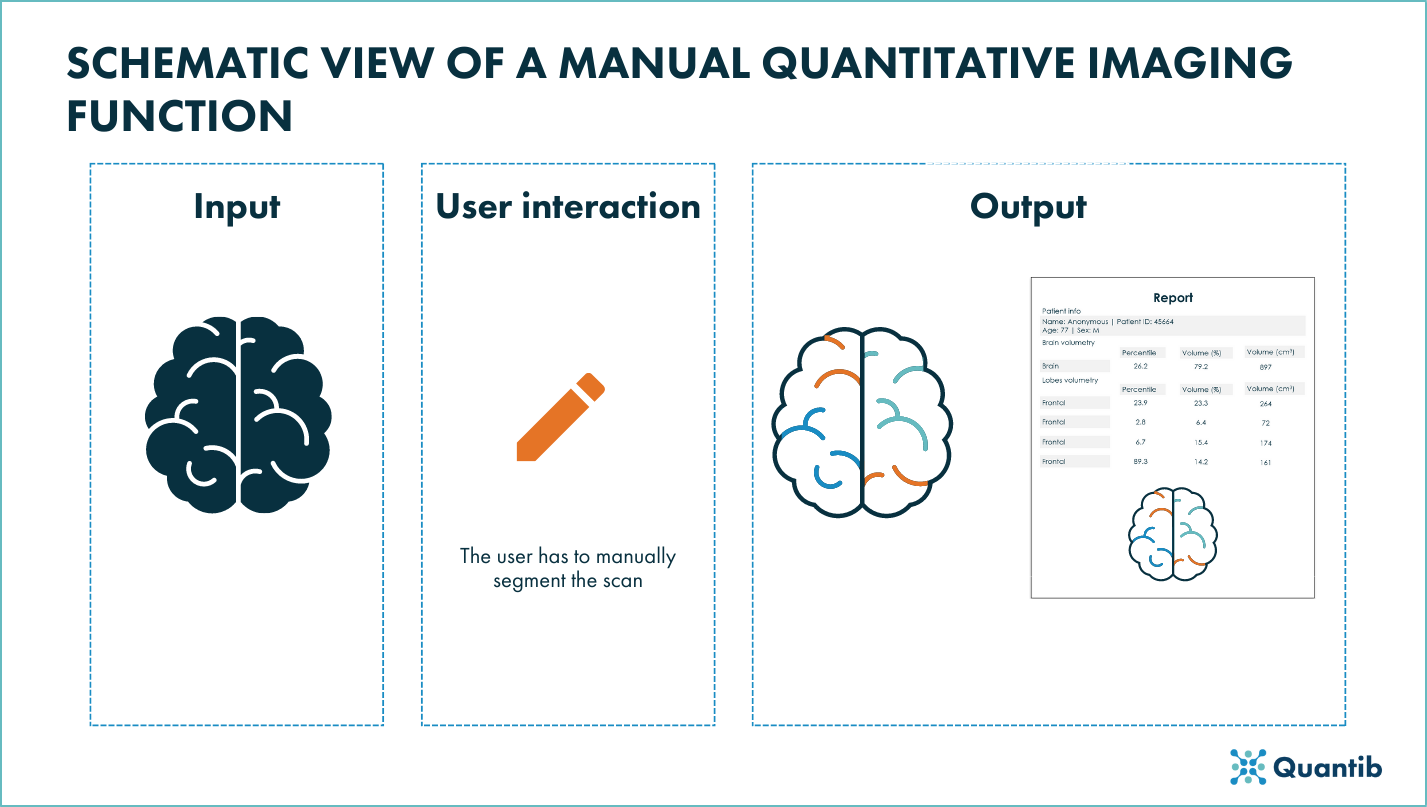
Figure 1: Schematic view of a manual quantitative imaging funciton of a radiological device.
Semi-automated quantitative imaging functions
For radiological devices that include a semi-automated quantitative imaging function the FDA recognizes that there might be “black box” steps that won’t be fully transparent to the user. Although the user, based on experience, might be able to mitigate gross errors derived of the function, there might still be some small errors. That’s why the FDA recommends manufacturers to do a more thorough assessment of the function’s performance, compared to a manual device.
The FDA guidance recommends1 the premarket submission for radiological devices with semi – automated quantitative imaging functions to include:
- Function description: it should describe functionality of the quantitative imaging function in depth and clearly specify if and what algorithms are implemented in the function. Besides that, it should also include which operations are to be performed by the user and which ones are to be performed by the quantitative imaging function.
- Technical Performance Assessment: it should include details about the software verification activities that ensure its correct implementation. Furthermore, it should also include data that verifies the performance specifications. Any claims based on performance over manual methods should be supported by studies that involve multiple use scenarios and multiple clinicians.
- Labeling (user interaction): it should include a clear description of the function’s functionality and the tasks that are to be performed by the user versus the imaging function, any limitations on input images and any performance specifications and its testing. It is also recommended to include the potential sources of error.

Figure 2: Schematic view of a semi-automated quantitative imaging funciton of a radiological device.
Fully automated quantitative imaging functions
When it comes to fully automated devices in which, by definition, there is no user interaction, the FDA considers that manufacturers should provide a “more robust analytical validation and more information describing the uncertainty associated with the output provided”1. For these devices, the FDA also recommends to exemplify instances in which the imaging function might give an incorrect output that is not easy to identify1.
- Function description: it should describe functionality of the quantitative imaging function in depth and clearly specify if and what algorithms are implemented in the function.
- Technical Performance Assessment:
- Labeling (user interaction): it should include a clear description of the function’s functionality, any limitations and any performance specifications and its testing. It is also recommended to include the potential sources of substantial error.
There are reporting guidelines for many different study designs, containing checklists of the points that the manuscript should, at least, contain.
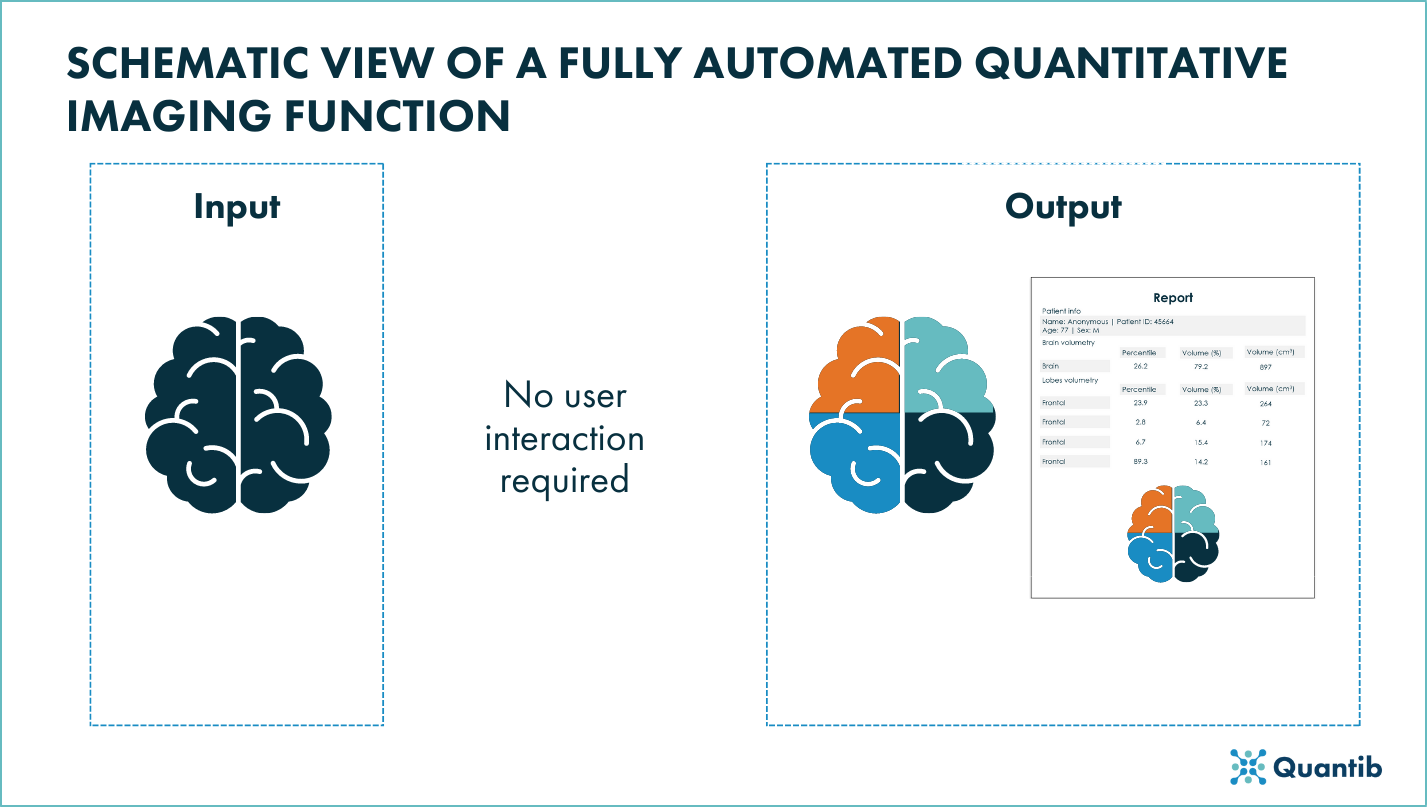
Figure 3: Schematic view of a fully automated quantitative imaging funciton of a radiological device.
Impact of the recommendations on end users
As you can see, premarket submissions require thorough documentation of the performance of quantitative imaging functions included in software as medical devices. With this Guidance1, the FDA shows their focus on the importance of software as a medical device, and how to ensure safe design and performance.
The Guidance1 states what information should be presented to the end user for them to use the device safe and as intended. Because interpreting quantitative imaging values is not always straight forward and there are many sources of error to take into account, together with their possible impact on the final values, this guidance also clarifies those aspects.
End users can now expect dedicated information on this to be present in the Information for Use, a separate document or during training sessions on the medical device. It is imperative to read and understand the quantitative imaging function’s intended use, intended situations, limitations on input data and operating range, to be able to have a good understanding of the actual meaning of the quantitative imaging values, and to make a proper interpretation of it.
The recommendation of having a rather detailed description of the algorithm and its performance available for end users also paves the way for a certain level of transparency, which facilitates comparison of different quantitative imaging algorithms and therefore selection of the most suitable devices for a particular use.
Impact of the recommendations on manufacturers
For manufacturers of quantitative imaging functions/devices, the recommendations of this guidance1 should not come as a complete surprise. Most recommendations can be considered good practice when developing and characterizing quantitative imaging functions. Having a list of topics to include in a premarket submission, together with a good set of definitions facilitates providing the desired level of documentation for the performance assessment.
Will the recommendations have an impact on Quantib and our way of working?
Our experience at Quantib is that, even though the guidance is recently published in its final version, our premarket submission reviews already were evaluated against most of the recommendations in this guidance, which was published as a draft with very similar content as back in 2019. At Quantib, we implemented the content of this draft guidance upon its publication, and therefore, we expect we will have to make minimal to no effort implementing the recommendations of the final guidance.
Conclusion
With the publication of the Guidance on Technical performance assessment of Quantitative imaging in radiological device premarket submissions1, the FDA defines its standard on how to perform and document a technical performance assessment, with increasing requirements for higher levels of automation of the quantitative imaging function.
The Guidance1 also sets recommendations on the information that should be included, which benefits the end user in safe and correct interpretation of quantitative imaging values.
Discover Quantib's FDA cleared AI-driven software for prostate MRI support and brain atrophy quantification.
Bibliography
-
Food and Drug Administration (FDA). Technical Performance Assessment of Quantitative Imaging in Radiological Device Premarket Submissions. https://www.regulations.gov. (2022).
-
Food and Drug Administration (FDA). Content of Premarket Submissions for Management of Cybersecurity in Medical Devices. http://www.regulations.gov. (2014).
-
Food and Drug Administration (FDA). Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices. http://www.fda.gov/MedicalDevices/ (2005).
-
Food and Drug Administration (FDA). Software as a Medical Device (SAMD): Clinical Evaluation. https://www.fda.gov/MedicalDevices/InternationalPrograms/IMDRF/default.htm. (2017).